


Introduction
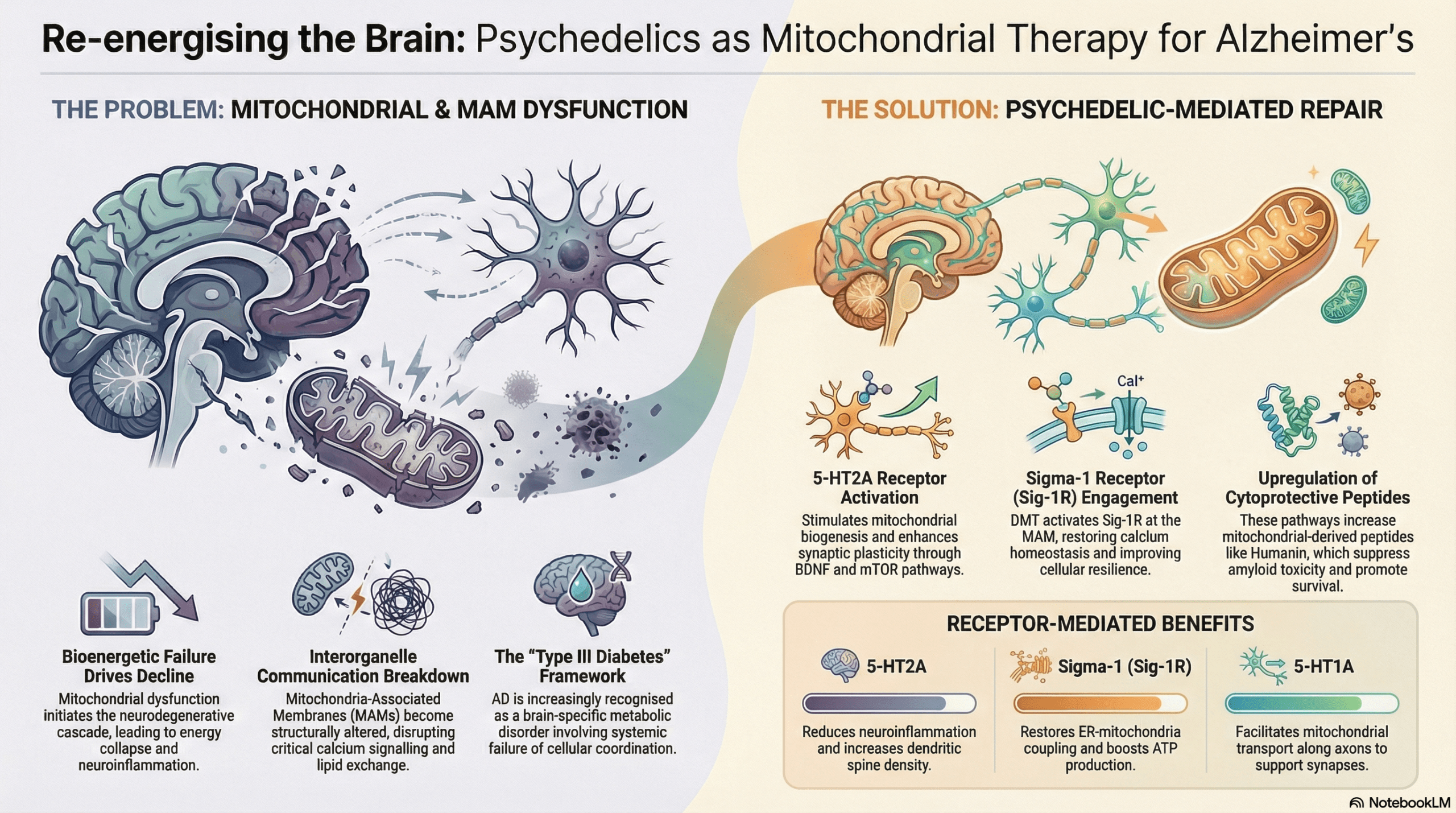
This high-stakes research addresses a critical therapeutic impasse, offering the Mitochondrial Cascade Hypothesis (MCH) as a more robust alternative to the limited success of the amyloid and tau hypotheses. The MCH repositions mitochondrial decline as the primary driver of neurodegeneration, characterizing Alzheimer’s as a state of “type III diabetes” where brain-specific insulin resistance leads to a total bioenergetic collapse. In this state, neurons effectively starve due to failing cellular powerhouses, regardless of glucose availability. The central research question explores whether classic psychedelics like psilocybin, LSD, DMT, and mescaline can intervene in this metabolic crisis to repair the fundamental power failure of the cell. This approach shifts the clinical focus from clearing protein aggregates to revitalizing the essential molecular machinery required for neuronal survival.
Infographic
Audio Summary
Key Findings
The research delineates a novel signaling axis where the activation of the Serotonin 2A (5-HT2A) and Sigma-1 (Sig-1R) receptors serves to restore mitochondrial integrity. A critical molecular event identified is the dissociation of Sig-1R from the ER-resident chaperone BiP (Binding immunoglobulin Protein). Upon binding ligands such as DMT, Sig-1R unmasks its active state and translocates to Mitochondria-Associated Membranes (MAMs). These MAMs serve as the essential hub where the endoplasmic reticulum and mitochondria “talk” through calcium exchange. By stabilizing IP3 receptors at these contact sites, DMT facilitates a calibrated calcium flux into the mitochondria, which is necessary to drive oxidative phosphorylation and boost ATP production.
Furthermore, the study highlights the role of the mitochondrial genome in conferring neuroprotection. The activation of Sig-1R promotes the upregulation of mitochondrial-derived peptides (MDPs), most notably Humanin (HN). This cytoprotective microprotein has demonstrated robust efficacy in neutralizing amyloid-induced toxicity and reducing tau hyperphosphorylation in preclinical models. These findings, observed in APP/PS1 mouse models and specialized cellular cultures, suggest that tryptaminic hallucinogens do not merely mask symptoms but may actively protect the mitochondrial proteome from pathological stress.
The anti-inflammatory profile of these compounds adds another layer of disease-modifying potential. Beyond suppressing IL-6 and TNF-alpha, the researchers highlight the inhibition of the NLRP3 inflammasome and the subsequent reduction of IL-1beta. This suppression of the chronic inflammatory cascade is vital for preserving synaptic architecture and preventing the metabolic exhaustion of microglia. While these molecular mechanisms provide a compelling framework for therapy, the authors emphasize that these results remain within the preclinical and hypothesis-generating scope, requiring rigorous validation before clinical application.
Conclusion
This study presents a measured and scientifically rigorous synthesis of the potential for hallucinogenic compounds to serve as regulators of interorganelle communication. While the restoration of mitochondrial biogenesis and MAM-mediated calcium signaling is mechanistically sound, the translational path is complicated by the unique vulnerability of the Alzheimer’s population. The potential for psychological distress and confusion at perceptually active doses necessitates a cautious approach to patient selection and neuropsychiatric safety. Future development may favor low-dose or non-hallucinogenic strategies that selectively engage the Sig-1R and 5-HT2A pathways to harness these bioenergetic benefits while minimizing the risk of adverse psychiatric outcomes in cognitively impaired patients.